Microscope technology development and the search for a free lunch
Posted by Alfred Millett-Sikking, on 10 July 2020
I fortuitously stumbled into the field of microscopy around 2013, just in time to enjoy the hype of the 2014 Chemistry Nobel Prize for super-resolved fluorescence imaging. As a newcomer, I was quite surprised that this nearly 400-year-old field could still catch the attention of Stockholm. After all, wasn’t a microscope just some lenses to magnify small objects? Little did I appreciate the complex train of devices and techniques that produce the cutting-edge microscopes of today! As an applied physicist and engineer, this provided a rich design and optimization space, with seemingly endless ways to innovate. I was delighted by the game of higher resolution, larger fields of view, faster frame rates, multidimensional imaging, and so on… but it was the overarching goal of microscopy that had so much appeal, which I put simply: to see more.
While learning about advanced microscopy techniques, it became apparent that gains in system performance in one area were often traded for losses somewhere else. Laser point scanning confocal microscopy (LCSM) [1] gave robust sectioning to produce excellent 3D images, but was typically slow and phototoxic. Multi-photon(MP) [2] and stimulated emission depletion (STED) [3] microscopy could greatly improve depth and resolution limitations, but with the drawbacks of point scanners, often increasing sample damage. Single molecule localization microscopy (SMLM), like (PALM) [4] or (STORM) [5], could yield an impressive boost in resolution, but at the expense of speed and complex sample preparation. Total internal reflection fluorescence (TIRF) [6] microscopy could give exquisite z resolution, but over a very limited range, and the structured illumination microscopy (SIM) [7] methods of the time were limited to shallow depths at moderate frame rates. So it seemed, quite reasonably, that design tradeoffs were inherent to the field, after all, there’s no free lunch.
It was in early 2014 that I became aware of Andrew York’s work on instant structured illumination microscopy (iSIM) [8]. This method elegantly combined multipoint confocal scanning with SIM in an all-optical ‘instant’ implementation, where images would arrive at the camera chip with a root(2) boost in resolution (a total 2-fold with post-processing). Fascinated by this idea I would spend the next year and a half designing, building and testing the first commercial prototype, the VT-iSIM (2015), and later enjoy seeing how Yokogawa [9] would incorporate the same method into their popular spinning disk platform, the SoRa (2018). Compared to other headline-grabbing super-resolution techniques this was a seemingly modest method, but with a salient punchline: it was a strict improvement in technology with no drawbacks.
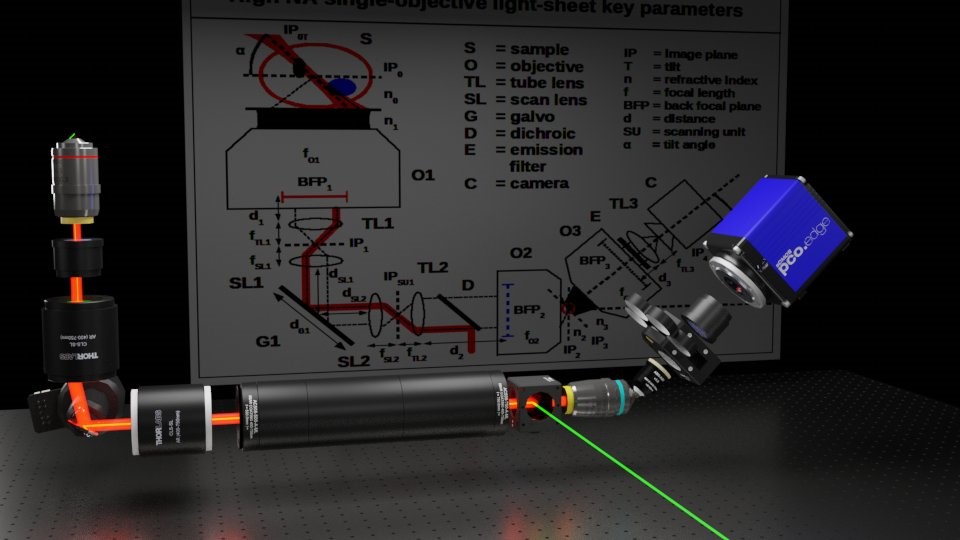
In the summer of 2015, I joined Andrew as a founding member of his new microscopy research group at Calico Life Sciences LLC in California. The company’s mission is to harness advanced technologies to increase our understanding of the biology that controls lifespan, and in this environment, we could continue our work on microscope technique development. In particular, we wanted to maintain the signature of iSIM and develop methods with wide utility and minimal shortcomings, a noble yet formidable goal. With ample support from Calico, the possibilities were somewhat overwhelming. What should we design and optimize? What are the rules and can we break them? What is possible and which microscopy method? We ask these questions regularly.
I have omitted thus far one of the simplest and most powerful concepts in biological imaging: selective plane illumination microscopy (SPIM) [10] or light-sheet fluorescence microscopy (LSFM) [11]. Fast, gentle and with good sectioning, the idea is as old as the 1925 Zigmondy Nobel Prize and has seen much attention in recent times with innovations like the DiSPIM [12] and Lattice (LSFM) [13], and polished commercial instruments, such as the Zeiss Lightsheet 7 and the Leica DLS. Yet despite its inherent advantages, LSFM has seen relatively little widespread adoption due to one simple (yet major) drawback: the interface.
The traditional coverslip ‘boundary’ defines where the biology ends and the engineering begins, it keeps samples sterile and allows the use of standard slides, dishes and multiwell plates (critical to many biological assays). Most light-sheet instruments violate this boundary by requiring two or more orthogonal lenses (for illumination and collection), and whilst some users can (and will) re-engineer their samples for a light-sheet microscope, most will not.
Andrew and I wrestled with light-sheet architecture during the early years at Calico, unsatisfied with the interface compromises we encountered in our multi-objective designs. We had deliberated over the brilliant oblique plane microscopy (OPM) invention of 2008 [14], where light-sheet excitation and emission were simultaneously passed through a single-objective. This underappreciated method resolved the boundary problem of typical light-sheets, but seemingly exchanged the advantage for heavy losses in resolution and optical efficiency. Even with this compromise, OPM had already been re-invented (and extensively used) in 2015 with the highly publicized SCAPE microscope [15], and would later be improved with the fast galvo scanning of SOPI (2018) [16]. Meanwhile Leica had licenced both the OPM and SCAPE technologies in 2016, a clear show of the appetite for a light-sheet in a convenient package.
Disheartened by the signal loss in OPM, we decided to evaluate the performance of the ingenious remote refocus (RR) technology [17] that underpinned the method, and even designed, built and tested our own high performance version. It was shortly after our RR publication in early 2018 that we chanced across the eSPIM [18] preprint on BioRxiv, which revealed a crucial insight: the major optical losses in OPM could be avoided.

Fueled by this new understanding, we set about re-designing the optical train for a new class of instrument, we called “A Bolt-on single-objective light-sheet design with uncompromised numerical aperture” (FOM 2019), later published in our “High NA single-objective light-sheet” (SOLS) article of 2019. From sample-to-camera, we eliminated the previous trade-offs and compressed the opto-mechanical difficulty into a new custom component: the AMS-AGY objective. This bold new lens, aka Snouty (and big brother KingSnout), now enable an extensive suite of design options with the speed, gentleness and sectioning of light-sheet and the convenience of a standard microscope interface, and for almost any fluorescence microscopy application.
We thank the many friends, colleagues and collaborators that helped us on this journey, and reflect on how the unique combination of our understanding of the technology, the support from Calico and the worthiness of the goal empowered us to make this breakthrough: a better microscope.

Alfred Millett-Sikking
Scientist, York Lab
Calico Life Sciences LLC,
South San Francisco, CA, 94010, USA
Reference list
1. Minsky, M. (1988). Memoir on inventing the confocal scanning microscope. Scanning 10: 128-138.
2. Denk, W., Stickler, J.H., Webb, W.W. (1990). Two-photon laser scanning fluorescence microscopy. Science 248: 73-76.
3. Hell, S.W., Wichmann, J. (1994). Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy. Optics Letter 19: 780-782.
4. Betzig, E., Patterson, G.H., Sougrat, R., Lindwasser, O.W., Olenych, S., Bonifacino, J.S., Davidson, M.W., Lippincott-Schwartz, J., Hess, H. (2006). Imaging intracellular fluorescent proteins at nanometer resolution. Science 313: 1642-1645.
5. Rust, M.J., Bates, M., Zhuang, X. (2006). Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nature Methods 3: 793-796.
6. Ambrose, E.J. (1956). A surface contact microscope for the study of cell movements. Nature 178: 1194.
7. Gustafsson, M.G.L. (2001). Surpassing the lateral resolution limit by factor of two using structured illumination microscopy. Journal of Microscopy 198: 82-87.
8. York, A.G., Chandris, P., Nogare, D.D., Head, J., Wawrzusin, P., Fischer, R.S. Chitnis, A., Shroff, H. (2013). Instant super-resolution imaging in live cells and embryos via analog image processing. Nature Methods 10: 1122-1126.
9. Azuma, T., Kei., T. (2015). Super-resolution spinning-disc confocal microscopy using optical photon reassignment. Optics Express 23: 15003-15011.
10. Huisken, J., Swoger, J., Del Bene, F., Wittbrodt, J., Stelzer, E.H.K. (2004). Optical sectioning deep inside live embryos by selective plane illumination microscopy. Science 305: 1007-1009.
11. Voie, A.H., Burns, D.H., Spelman, F.A. (1993). Orthogonal-plane fluorescence optical sectioning: Three-dimensional imaging of macroscopic biological specimens. Journal of Microscopy 170: 229-236.
12. Wu, Y., Wawrzusin, P., Senseney, J., Fischer, R.S., Christensen, R., Santella, A., York, A.G., Winter, P.W., Waterman, C.M., Bao, Z., Colon-Ramos, D.A., McAuliffe, M., Shroff, H. (2013). Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nature Biotechnology 31: 1032-1038.
13. Chen, B-C., Legant, W.R., Wang, K., Shao, L., Milkie, D.E., Davidson, M.W., Janetopoulos, C., Wu, X.S., Hammer III, J.A., Liu, Z., English, B.P., Mimori-Kiyosue, Y., Pomero, D.P., Ritter, A.T., Lippincott-Schwartz, J., Fritz-Laylin, L., Mullins, R.D., Mitchell, D.M., Bambenek, J.N., Reymann, A-C., Boehme, R., Grill, S.W., Wang, J.T., Seydoux, G., Tulu, U.S., Kiehart, D.P., Betzig, E. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346: 1257998.
14. Dunsby, C. (2008). Optically sectioned imaging by oblique plane microscopy. Optics Express 16: 20306-20316.
15. Bouchard, M.B., Voleti, V., Mendes, C.S., Lacefield, C., Grueber, W.B., Mann, R.S., Bruno, R.M., Hillman, E.M.C. (2015). Swept confocally-aligned planar excitation (SCAPE) microscopy for high speed volumetric imaging of behaving organisms. Nature Photonics 9: 113-119.
16. Kumar, M., Kishore, S., Nasenbeny, J., McLean, D.L., Kozorovitskiy, Y. (2018). Integrated one- and two-photon scanned oblique plane illumination (SOPi) microscopy for rapid volumetric imaging. Optics Express 26: 13027-13041.
17. Botcherby, E.J., Juskaitis, R., Booth, M.J., Wilson, T. (2008). An optical technique for remote focusing in microscopy. Optics Communications 281: 880-887.
18. Yang, B., Wang, Y., Feng, S., Pessino, V., Stuurman, N., Huang, B. (2018). High numerical aperture Epi-illumination selective plane illumination microscopy. bioRxiv 273359
 (2 votes, average: 1.00 out of 1)
(2 votes, average: 1.00 out of 1)Get involved
Create an account or log in to post your story on FocalPlane.
More posts like this

Filter by
- NewsApply
- DiscussionsApply
- How toApply
- ToolsApply
- Case studiesApply
- InterviewsApply
- JobsApply
- EducationApply
- Blog seriesApply
- Latin America Bioima..gingApply
- From Zero to Qupath ..HeroApply
- Asian Microscopists ..and Cell BiologistsApply
- AIC at HHMI JaneliaApply
- Deep Learning for Bi..o-image analysisApply
- GloBIAS – updates fr..om the communityApply
- WAMBIAN: West Africa.. in FocusApply
- Volume EMApply
- Latin American Micro..scopistsApply
- Bio-image Analysis w..ith NapariApply
- Imaging with…Apply
- Towards Global Acces..sApply
- Highlights from Euro..-BioImagingApply
- LSFM seriesApply
- DIY MicroscopyApply
- View all