Structural Repetition Detector (SReD): quantitative mapping of molecular complexes through microscopy
Posted by Afonso Mendes, on 23 September 2024
Unbiased, multi-dimensional, multi-scale and GPU-accelerated analysis of structural patterns across all microscopy modalities
From biomolecules to larger assemblies and cellular architectures, molecular structures govern biological processes. Identifying these repetitive patterns is essential to understand their functions and the underlying biological mechanisms. While microscopy offers molecular-level detail, manually detecting structural motifs is labor-intensive, susceptible to bias, and requires specialised expertise.
The Structural Repetition Detector (SReD – reads as “Shred”!) offers a powerful solution for uncovering structural repetition in microscopy data without the need for prior knowledge of the structures. It provides an objective, quantitative approach to structure detection by scoring patterns based on their frequency. Unlike other methods that rely on user-defined templates, manual segmentation or training data, SReD works directly on raw image data, ensuring unbiased analysis. Its easy-to-use interface makes it accessible to researchers without programming experience.
We released SReD as an ImageJ and Fiji plugin that harnesses GPU processing to enable unbiased structure detection and quantitative analysis in microscopy images.
You can find a detailed description with examples in real microscopy datasets in the SReD preprint1.
Head onto the SReD Github for installation instructions and tutorials!

Key features
– Unbiased structure detection and quantitative analysis.
– GPU acceleration to mitigate computational burden.
– 3D, time and multi-scale capabilities.
– Generalisable to all microscopy modalities.
What can SReD do?
SReD offers a solution to structure detection challenges by identifying repetitive patterns in microscopy data without requiring prior information. It analyses structural patterns based on their repetition, enabling robust quantitative analysis. Compatible with all microscopy data, SReD operates directly on image reconstructions and is user-friendly, requiring no programming experience.
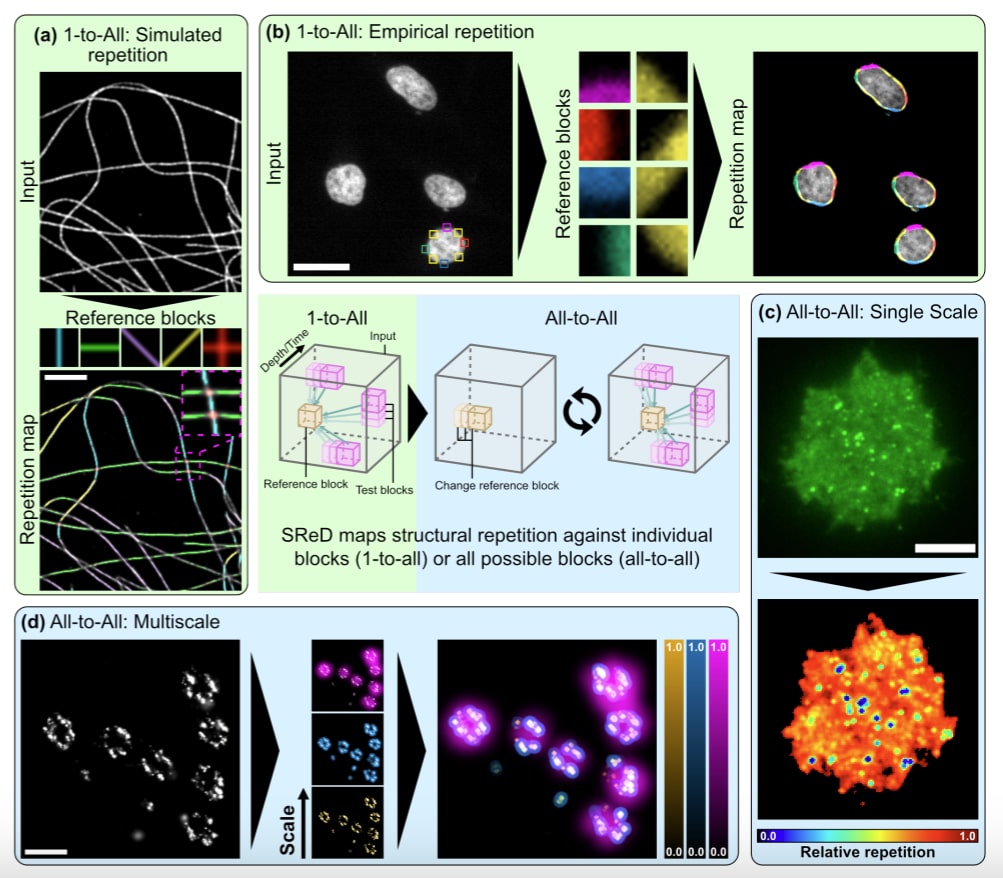
SReD is tailored for biologists, and its applicability was demonstrated across various biological contexts. From microtubules, to nuclear pore complexes and viruses, SReD provided valuable insights into structural patterns present in datasets.
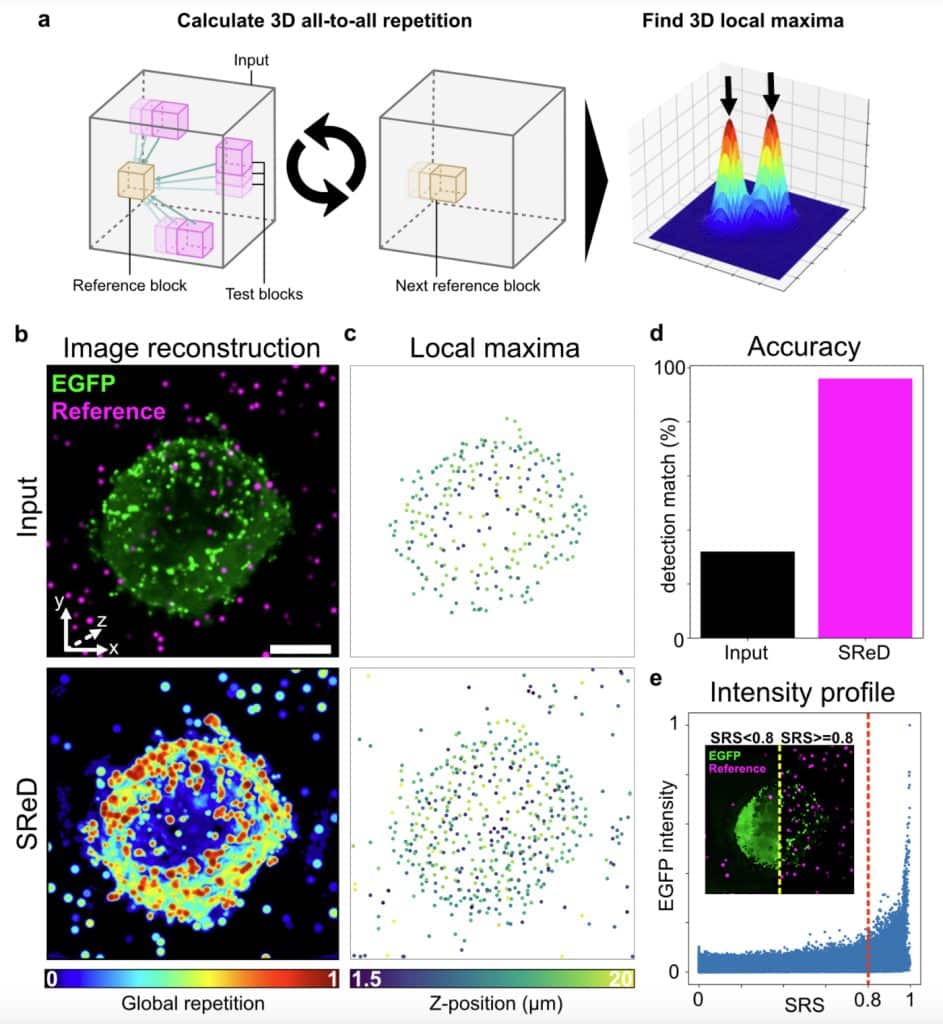
Sneak-peek showcase: Detection of HIV-1 Gag particles
We used SReD’s “all-to-all” sampling scheme to detect HIV-1 Gag-EGFP structures in 3D. Virus-like particles (VLPs) were identified from both the input data and SReD’s repetition maps by calculating local maxima. Artificially generated particles served as reference points for accuracy evaluation. The repetition maps significantly outperformed the input data for VLP detection, and by scoring patterns based on frequency, SReD differentiated structurally relevant regions from the EGFP background.
🔍🤓 Read ahead for more on the story behind SReD! 🤓🔎
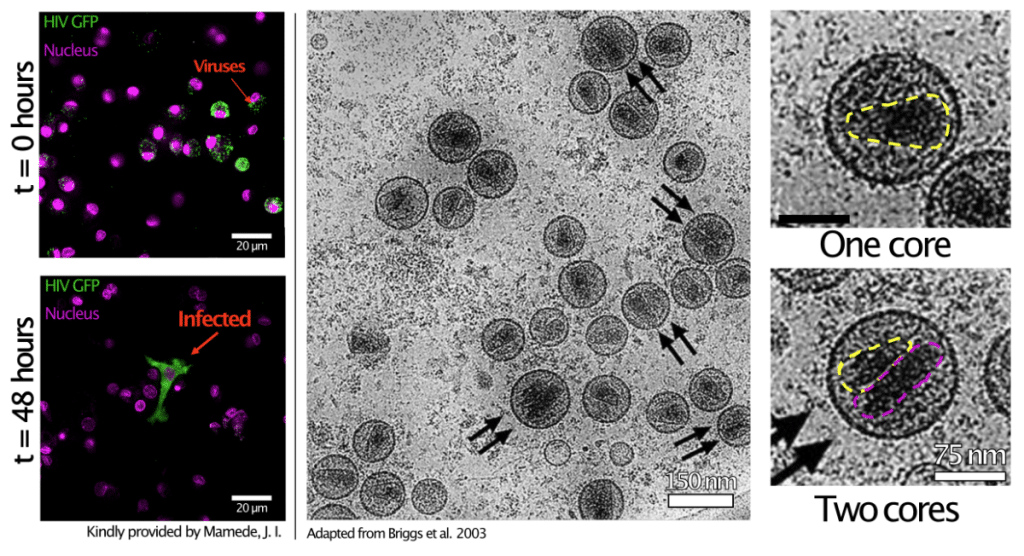
The problem of not knowing what you’re looking for
About 5 years ago, I was struck by an intriguing observation: despite its high prevalence, HIV-1 is surprisingly inefficient at establishing infection. This paradox, known among experimentalists but absent in the literature, raised questions. Briggs et al.2 had reported that about a third of viral particles contained two cores instead of one, and I hypothesised that this structural variability could influence infectivity. I set out to explore viral heterogeneity during assembly using fluorescence microscopy but soon realised that these structures had never been characterised before, leaving me without a reference for what to expect.
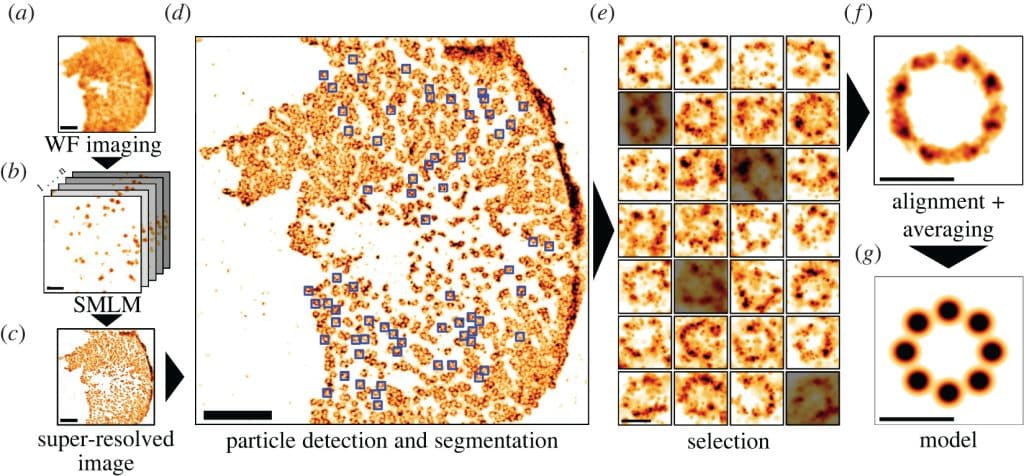
Template bias, averaging variability and frying cells with lasers
My project required Single Particle Analysis (SPA), which analyses the morphology of single structures (named “particles”) to create a structural map. Upon extensively reviewing existing methods3, I noted two key drawbacks: the need for a user-provided template, leading to bias, and limited sensitivity to structural variability due to averaging. Heydarian et al.4,5,6 developed a template-free approach that compared each structure with others, allowing for variability characterisation, but still required manual segmentation and relied on localisation data, which wasn’t suited for long-term live-cell microscopy.
Converging ideas
Moving away from localisation data meant using image reconstructions from more live-cell-friendly microscopy techniques. This pointed us towards correlative intensity analysis. Based on this, we developed a low-level approach for unbiased structure detection, based on the idea that structures appear as local textures. By comparing a block from the image with all others, we could detect repetitions and score their likelihood, with the average score reflecting estimated frequency. Extending this approach using the entire universe of image blocks as a reference resulted in unbiased analysis. This method enabled quantitative characterisation of structurally relevant regions without prior information, using the full set of blocks extracted from the image.
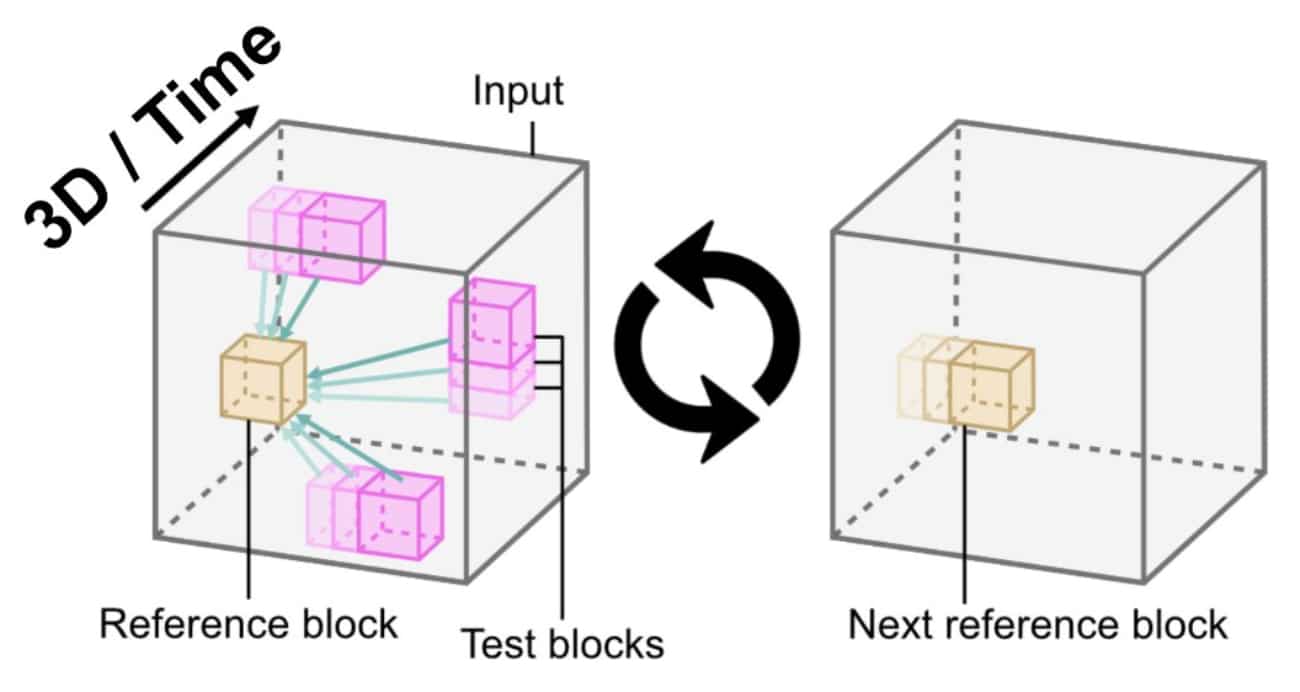
Releasing SReD for everyone
We soon realised that unbiased structure detection and analysis was a widespread challenge. This, combined with our passion for open science, inspired us to develop SReD as an open-source ImageJ and Fiji plugin. We ensured that others could use it without restrictions and demonstrated its applicability in various common biological contexts beyond our own.
👇 The video below shows how to get started with using SReD 👇
Additionally, the SReD Github page has plenty of information, including step-by-step tutorials that will guide you through SReD’s capabilities.
We are constantly looking into ways to improve our software. This means that we are 100% open to feedback and suggestions.
References
1 – Mendes, A. et al. (2024) Structural Repetition Detector: multi-scale quantitative mapping of molecular complexes through microscopy. Biorxiv. DOI: https://doi.org/10.1101/2024.09.16.613204.
2 – Briggs, J. A. G. et al. (2003) Structural organization of authentic, mature HIV-1 virions and cores. Embo J. 22(7):1707–1715. DOI: https://doi.org/10.1093/embojcdg143.
3 – Mendes, A. et al. (2022) Mapping molecular complexes with super-resolution microscopy and single-particle analysis. Open Biology. 12(7). DOI: https://doi.org/10.1098/rsob.220079.
4- Hamidreza et al. (2018) Template-free 2D particle fusion in localization microscopy. Nat Met. 15: 781–784. DOI: https://doi.org/10.1038/s41592-018-0136-6.
5 – Hamidreza et al. (2021) 3D particle averaging and detection of macromolecular symmetry in localization microscopy. Nat Comms. 12(2847). DOI: https://doi.org/10.1038/s41467-021-22006-5.
6 – Huijben, T. A. P. M. (2021) Detecting structural heterogeneity in single-molecule localization microscopy data. Nat Comms. 12(3791). DOI: https://doi.org/10.1038/s41467-021-24106-8.
 (1 votes, average: 1.00 out of 1)
(1 votes, average: 1.00 out of 1)Get involved
Create an account or log in to post your story on FocalPlane.
More posts like this


Filter by
- NewsApply
- DiscussionsApply
- How toApply
- ToolsApply
- Case studiesApply
- InterviewsApply
- JobsApply
- EducationApply
- Blog seriesApply
- Volume EMApply
- Latin American Micro..scopistsApply
- Bio-image Analysis w..ith NapariApply
- Imaging with…Apply
- Towards Global Acces..sApply
- Latin America Bioima..gingApply
- From Zero to Qupath ..HeroApply
- Asian Microscopists ..and Cell BiologistsApply
- AIC at HHMI JaneliaApply
- Deep Learning for Bi..o-image analysisApply
- GloBIAS – updates fr..om the communityApply
- Highlights from Euro..-BioImagingApply
- LSFM seriesApply
- DIY MicroscopyApply
- View all