Imaging spotlight: Cryo-ExM reveals the architecture of the human cytotoxic immunological synapse
Posted by FocalPlane, on 15 June 2026
In this behind the paper story, Benita Wolf and colleagues share their research using cryo-expansion microscopy (cryo-ExM) to map the architecture of the human cytotoxic immunological synapse.
The immunological synapse is one of the most spatially organised structures in cell biology. When a cytotoxic T cell engages its target, it polarises within seconds: receptors cluster, the centrosome translocates to the contact site, and lytic granules are delivered to a precise location on the plasma membrane. We wanted to look at this architecture in human T cells at a level of detail that conventional approaches could not reach — and to do it without the fixation artefacts that have complicated synapse imaging for decades.
What are the key results from your paper?
We applied cryo-expansion microscopy (cryo-ExM) to primary human T cells and used it to resolve their three-dimensional architecture at a lateral resolution below 70 nm. The combination of millisecond cryo-fixation by plunge-freezing and isotropic physical expansion gave us access to features that have been very difficult to study faithfully with conventional approaches: the actin and microtubule cytoskeleton in the same cell, fragile membrane structures including microvilli and tunneling nanotubes, the molecular architecture of centrioles at the synapse, and the internal organization of intact lytic granules.
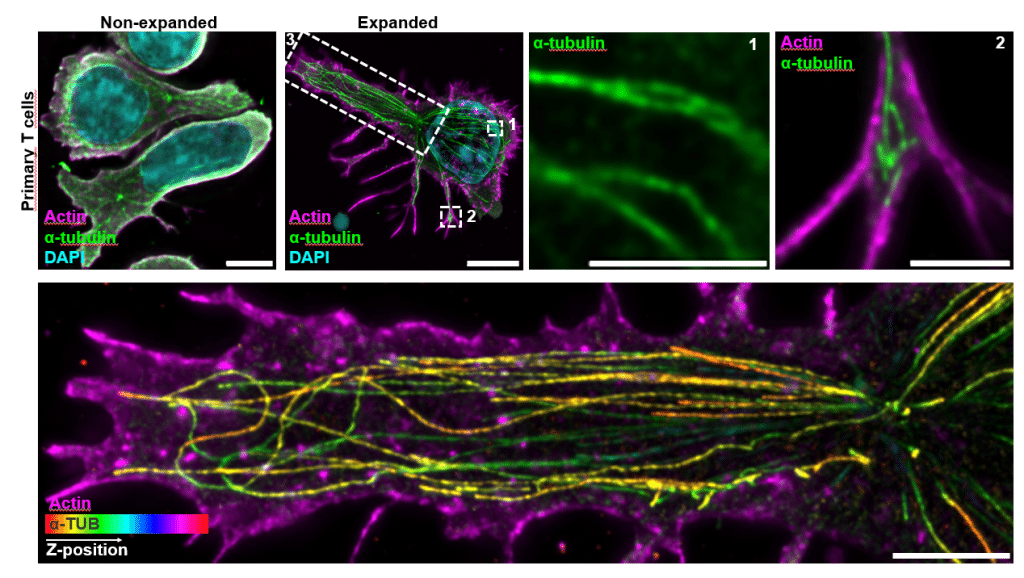
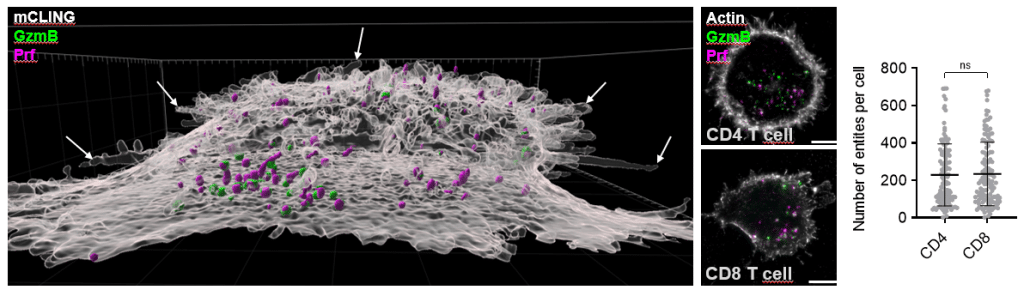
Three findings stood out for us. First, we discovered an adhesion-dependent, dome-like membrane architecture beneath activated T cells, which collapses upon ICAM-1 engagement. This suggests that T cells are intrinsically capable of generating a concave synaptic topology under activation alone, rather than this geometry being imposed solely by the target cell. Second, we directly visualized single-core and multi-core lytic granules in primary human CD4 and CD8 T cells, including the first intracellular evidence of perforin organization within multi-core granules in human cells. We could map LAMP1, granzyme B, perforin, and the trafficking machinery (RAB7A, RAB27A, UNC13D, SYTL2, VAMP7, STX11, TSP1) within individual granules. Third, perhaps the most clinically translatable finding, we adapted expansion microscopy to FFPE human glioblastoma and melanoma brain metastasis tissue, enabling nanoscale visualization of tumor-infiltrating T cells and their lytic content directly in archived patient samples.
A broader biological headline, surprising to us, is that CD4 and CD8 T cells are equipped with a remarkably similar cytotoxic machinery — comparable numbers of lytic entities, similar granule architectures, and when CD4 T cells are equipped with a tumor-specific TCR, comparable serial killing capacity against tumor targets.
The technical headline is that cryo-ExM is now a workable tool for human T cell biology, opening up questions that have been stuck for years.
Which imaging and image analysis techniques did you use in your research, and why did you use cryo-fixation rather than chemical fixation?
We imaged using a Leica DMi8 Thunder widefield microscope and a Leica Stellaris 8 FALCON confocal microscope. For analysis, we used Fiji (ImageJ) with custom macros — for centriole measurements, synaptic morphometry, concentric bin analysis of protein distribution at the synapse, and 3D segmentation of lytic granules — and Imaris 10.1 for 3D surface reconstructions. For lytic granule segmentation, we developed a machine learning-assisted pipeline using the LABKIT plugin in Fiji, trained on manually annotated 3D stacks, which allowed us to automatically quantify granule number, volume, calculated diameter, spatial position, and protein content across hundreds of cells. All custom macros are openly available on GitHub.
Cryo-fixation by plunge-freezing into liquid ethane was a deliberate choice over conventional chemical fixation, for several reasons:
i) Chemical fixatives like paraformaldehyde are convenient but introduce a known set of artefacts that are particularly problematic for immune cells. They differentially preserve actin and microtubules, which is a real limitation when the central question concerns coordinated cytoskeletal remodelling at the synapse. They can disrupt membrane topology, induce granule fusion artefacts, and shrink samples during dehydration. They also extract soluble granule cargo and mask antibody epitopes — especially relevant for granzymes and perforin, which are notoriously difficult to preserve and stain after chemical fixation.
ii) Cryo-fixation, by contrast, preserves the cell in milliseconds in its native hydrated state, giving us a much more faithful starting point. Combined with expansion microscopy via freeze substitution, we get isotropic ~4-fold physical magnification of structures that can then be imaged on conventional widefield or confocal systems, with effective resolutions approaching those delivered by super-resolution methods — but applied to a near-native cellular state, with multicolour flexibility, and across whole cells in three dimensions. This combination occupies a niche that none of the existing fixed super-resolution methods (STED, dSTORM, DNA-PAINT) can really fill: nanoscale, volumetric, multicolour, near-native, on standard fluorescence microscopes.
Do you have any technical tips or challenges that you can share?
A few things we learned the hard way that might save others time:
The plunge-freezing step is the single most important determinant of downstream quality. Variability here propagates into everything else, so it is worth investing in a reliable plunger and standardising the blotting and humidity conditions before scaling up. We designed a tweezer holder to keep the sample aligned with the centre of the cryo-chamber containing the liquid ethane — small adjustments like this matter for reproducibility.
Cell type matters more than the protocols sometimes acknowledge. Primary human T cells behave differently from immortalised lines or murine cells in terms of how they tolerate the expansion chemistry. We had to invest substantial piloting work to ensure isotropic expansion, and we routinely verify isotropy using the nuclear cross section before and after expansion as an internal control. Researchers moving from one cell system to another should expect to re-optimise rather than transfer protocols directly.
Antibody choice and staining order are non-trivial. Some epitopes survive expansion well, others do not. We always stained post-expansion to maximise labelling efficiency. We worked through our epitopes of interest empirically and would recommend pilot panels before committing to a full experimental series. This is particularly true for granule cargo (granzyme B, perforin) and trafficking proteins.
For tissue work, we found that FFPE samples could be expanded with a tissue-adapted U-ExM protocol, but the deparaffinization, crosslinking prevention, and gelation steps require careful temperature control. The ability to perform multiple rounds of post-expansion immunofluorescence staining and stripping was important for multiplexing on the same sample.
Beware of subtle distortions. Physical expansion can introduce local anisotropy in the gel matrix, and cryo-fixation can cause ice crystal formation if water vitrification is incomplete. Such defects are usually readily recognisable visually and can be excluded from analysis, but they reinforce the need to interpret geometric measurements comparatively rather than as exact absolute physical dimensions.
Are there any advances in imaging or analysis that would help your research going forward?
Several directions excite us:
Cryo-ExM directly in fresh tissue is the next major step. Achieving true near-native preservation in tissue would require rapid cryo-immobilization by high-pressure freezing of thin specimens (~100–300 μm) without prior chemical fixation, followed by freeze substitution and expansion. This would open the door to nanoscale, three-dimensional visualization of immune synapses, lytic granules, and cellular interfaces directly in patient-derived samples — bridging the long-standing gap between high-resolution structural cell biology and in vivo human immunology. In tumour tissues specifically, this could be transformative for dissecting mechanisms of immune evasion in situ.
Faster and gentler post-expansion imaging would help us substantially. Expansion produces large data volumes (4-fold expansion in each dimension means ~64-fold more voxels), and current acquisition times limit how many conditions and cells we can sample. Improvements in light-sheet imaging of expanded gels, in adaptive optics, and in computational denoising would all help.
Better tools for handling and analysing large 3D datasets are essential. We invested substantial effort into custom Fiji macros and machine learning pipelines for our granule analysis, but the broader community would benefit from standardised, well-documented frameworks for segmenting and quantifying nanoscale structures in expanded cells. Validation tools for expansion isotropy at the local (organelle-scale) level would also strengthen quantitative rigour.
Finally, improved correlation between cryo-ExM and live-cell imaging modalities would be enormously valuable. Cryo-ExM captures static snapshots; live-cell modalities capture dynamics. Tools that allow the same cell to be imaged dynamically, then cryo-fixed and expanded for nanoscale structural analysis, would close one of the most important gaps in our current toolkit.
 (No Ratings Yet)
(No Ratings Yet)Get involved
Create an account or log in to post your story on FocalPlane.
More posts like this


Filter by
- NewsApply
- DiscussionsApply
- How toApply
- ToolsApply
- Case studiesApply
- InterviewsApply
- JobsApply
- EducationApply
- Blog seriesApply
- Volume EMApply
- Latin American Micro..scopistsApply
- Bio-image Analysis w..ith NapariApply
- Imaging with…Apply
- Towards Global Acces..sApply
- Latin America Bioima..gingApply
- From Zero to Qupath ..HeroApply
- Asian Microscopists ..and Cell BiologistsApply
- AIC at HHMI JaneliaApply
- Deep Learning for Bi..o-image analysisApply
- GloBIAS – updates fr..om the communityApply
- WAMBIAN: West Africa.. in FocusApply
- Highlights from Euro..-BioImagingApply
- LSFM seriesApply
- DIY MicroscopyApply
- View all